The count matrix is managed as a UnimodalData object defined in PegasusIO module, and users can manipulate the data from top level via MultimodalData structure, which can contain multiple UnimodalData objects as members.
For this example, as show above, data is a MultimodalData object, with only one UnimodalData member of key "GRCh38-rna", which is its default UnimodalData. Any operation on data will be applied to this default UnimodalData object.
UnimodalData has the following structure:
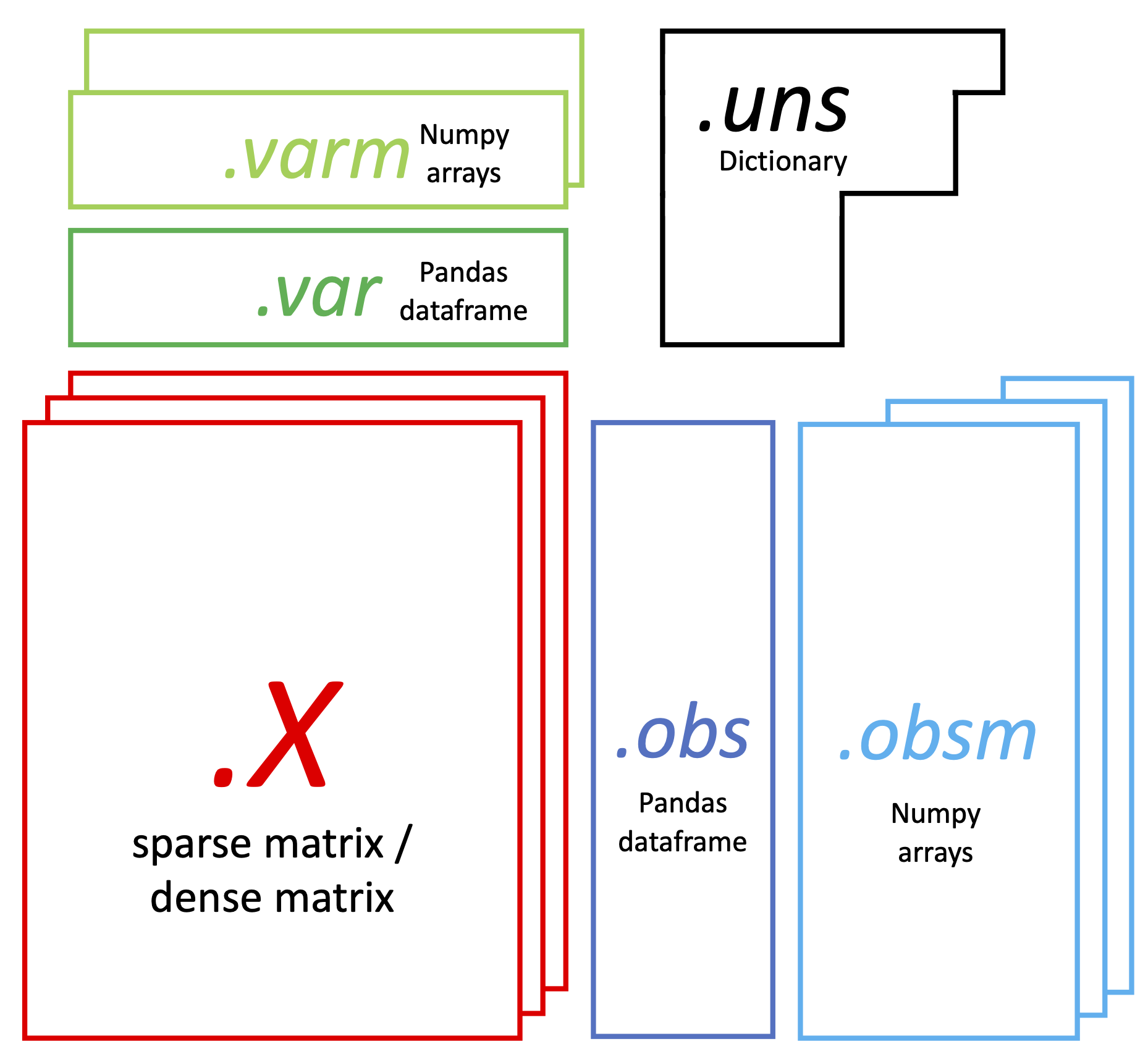
It has 6 major parts:
- Raw count matrix:
data.X, a Scipy sparse matrix (sometimes can be a dense matrix), with rows the cell barcodes, columns the genes/features:












